Alignment methods (Part 1)
Previous class check-up
- We are familiar with git/github
- The class notes are in its own repo:
git cloneto downloadgit pullto update them, or- fork and
git pull upstream master
- The class notes are in its own repo:
- We understand the importance of reproducible scripts
- We created a github repo for the class project (remember to add me as collaborator to your repo)
Learn@Home: Sequencing check-up
- We understand sequencing technologies
- We start thinking about data for the class project -> beginning of work from home in your data!
Learning objectives
At the end of today’s session, you
- will be able to explain the most widely used algorithms for pairwise sequence alignment (Needleman-Wunsch algorithm)
Pre-class work
None.
Stop and check
After today, we will assume that 1) we are all comfortable with the terminal and 2) we are all comfortable with git/github. Note that no one needs to memorize anything. You will always have access to the notes and google. If you are still struggling with the terminal and/or git/github, please check out the resources at the end of the Reproducibility notes. Let’s do this short quiz.
Ask ChatGPT
This is a ChatGPT-friendly class! Use ChatGPT to troubleshoot computer issues (keep in mind it sometimes lie).
What is multiple sequence alignment (MSA)?
- We infer homology when sequences are evolutionarily related: that is, when they descend from a common ancestor
- Intuitively, an MSA method inserts gap characters (
-) inside input sequences to produce a set of longer sequences that are all of the same length, such that residues at the same position in different sequences (aligned residues) share some common properties (homology) - MSA is a crucial step since phylogenetic inference methods assume that residue homology relationships are correctly reflected by the input sequences
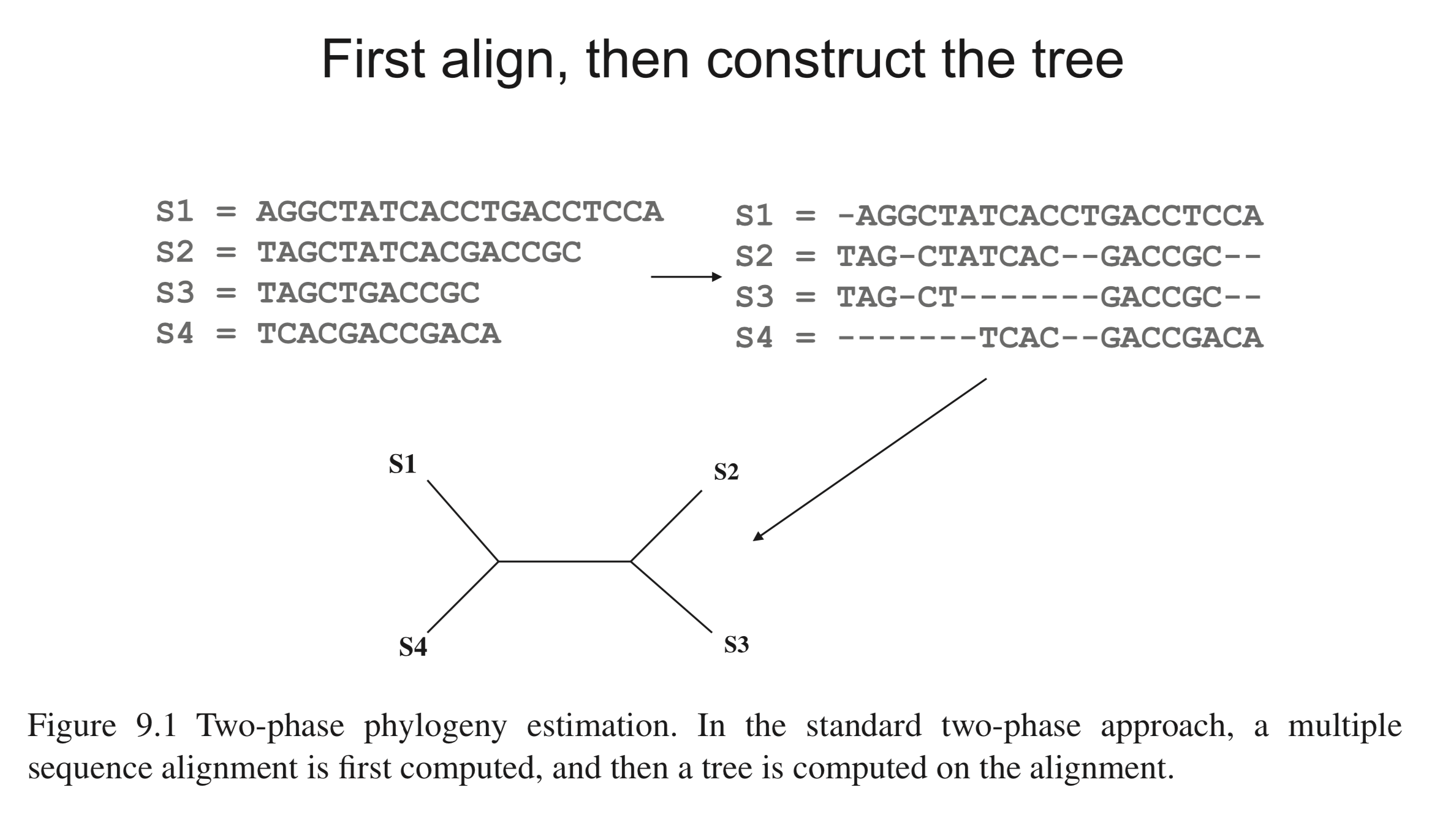
Figure 9.1 in Warnow (2018) Computational phylogenetics
The true MSA reflects the historical substitution, insertion and deletion evolutionary events:
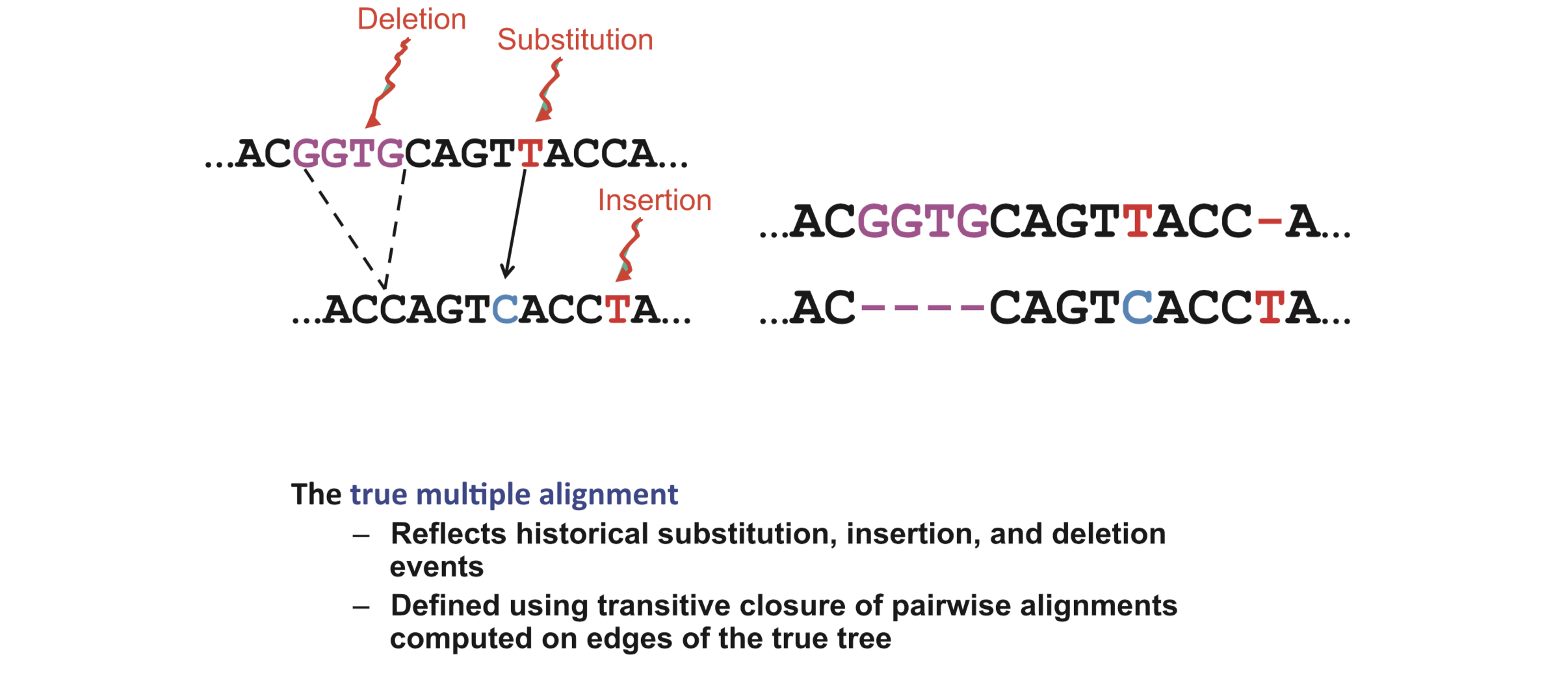
Figure 9.2 in Warnow (2018) Computational phylogenetics
Why is it one of the most computationally intensive tasks? Alignment can be not identifiable or unique.
Example 9.1
Suppose that we knew the true evolutionary events from sequence ACAT to AGAT. Suppose that we knew that it was not a substitution, but a deletion of C (thus creating AAT) followed by an insersion of G. Even in this scenario (when we know the truth), there are two ways to represent the alignment:
AC-AT
A-GAT
and
A-CAT
AG-AT
In-class activities
Table 9.1 (Warnow). What would be the alignment of the sequence S=ACATTA which evolves into S'=TACA if we knew the true evolutionary events?
- deletion of the first two nucleotides
AC - deletion of the second
T - substitution of
TintoC - insersion of
Tat the front
Solution: In class.
Table 9.2 (Warnow). Without knowing the true evolutionary events from S=ACATTA to S'=TACA, what would you think is a good alignment?
Solution: You probably choose an alignment where none (or few) of the true homology relationships are correct.
- The true alignment has 4 events: 2 deletions, 1 insersion, 1 substitution
- The estimated alignment we created now also has 4 events: 3 deletions and 1 insersion
- The algorithm will ultimately choose an alignment based on how we penalize each of the events
First key insight for MSA: We are guiding the algorithms by selecting the penalties for evolutionary events: substitutions, deletions, insersions. It has nothing to do with true evolutionary events or true homology.
MSA algorithm
Input: unaligned sequences (different lengths)
Output: aligned sequences (same length) where each site is an assertion of homology
Steps in MSA:
- Define cost of each event: deletion, insertion, substitution
- Learn to obtain the optimal pairwise alignment with the minimum cost (“edit distance”)
- Learn to obtain the optimal multiple sequence alignment: we need to be able to align alignments
1. Cost of evolutionary events
- cost of deletion/insersion (cost of gap): 1
- cost of substitution: 1
Some software/books will use “weights” instead of costs: weight for match: 5; weight for gap: -1; weight for mismatch (substitution): -1. I prefer to use costs because the idea of negative weights is not intuitive.
In-class activities
Table 9.3 (Warnow). How would you align the sequences S=AACT and S'=CTGG when:
- cost of gap: 1
- cost of substitution: 3 ?
Table 9.3 (Warnow). How would you change the alignment between sequences S=AACT and S'=CTGG if the costs were:
- cost of gap: 4
- cost of substitution: 1
Solution: In class.
Other types of costs
There are other ways to measure cost/penalty:
- sequence identity: number of identical sites in an alignment divided by the total number of aligned positions
- biochemical similarity when assigning costs of substitutions (Figure 3.4 HB 3): PAM weight matrices and BLOSUM62 weight matrices

We will continue to use cost for simplicity.
2. Pairwise sequence alignment
- Pairwise alignment of short toy sequences (like in the examples) can be done by hand
- Pairwise alignment of real sequences would be too difficult to do by hand, so we need smart algorithms: Needleman-Wunsch algorithm
- Needleman-Wunsch is a dynamic programming algorithm
- Dynamic programming: optimization algorithm that simplifies a complicated problem by breaking it down into simpler sub-problems in a recursive manner
Needleman-Wunsch algorithm
- Ingredients:
- Two sequences: \(A=a_1 a_2 ...a_m\) and \(B=b_1 b_2 ...b_n\)
- 1) cost of gap and 2) cost of substitution
We will define a matrix $F$ whose entries $F(i,j)$ will represent the minimum cost to align sub-sequences $A_i$ and $B_j$ based on the costs.
Stop and check
Do you know what a matrix is and how (i,j) entries are defined?
Main principle: When we want to compute $F(i,j)$, we assume that we have already computed all smaller sequences (sub-problems): $F(i-1,j-1), F(i,j-1), F(i-1,j)$.
The algorithm will focus on two residues at a time: $a_i$ and $b_j$, and for them, they are three options:
- $a_i$ and $b_j$ are aligned together in the final site. Then the other sites define a pairwise alignment of $A_{i-1}$ and $B_{j-1}$
- $a_i$ is aligned with a gap in the final site. Then $A_{i-1}$ and $B_{j}$ defined a pairwise alignment
- $b_j$ is aligned with a gap in the final site. Then $A_{i}$ and $B_{j-1}$ defined a pairwise alignment
Example of notation
Let $A=a_1 a_2 a_3 a_4 a_5 a_6$ and $B=b_1 b_2 b_3 b_4 b_5$, and suppose you want to align sites $a_5$ ( $i=5$ ) and $b_3$ ( $j=3$ ). We have three options:
- $a_5$ and $b_3$ are aligned together in the final site. Then the other sites define a pairwise alignment of $A_4=a_1 a_2 a_3 a_4$ and $B_2=b_1 b_2$ (we do not know how at this point)
- $a_5$ is aligned with a gap in the final site. Then $A_4=a_1 a_2 a_3 a_4$ and $B_3=b_1 b_2 b_3$ define a pairwise alignment
- $b_3$ is aligned with a gap in the final site. Then $A_5=a_1 a_2 a_3 a_4 a_5$ and $B_2=b_1 b_2$ define a pairwise alignment
Needleman-Wunsch algorithm: Costs
- If $a_i$ and $b_j$ are aligned together, then the cost is 0 if $a_i=b_j$ and 1 (or whatever cost of substitution defined) if they are different. Hence, the total cost is $F(i,j) = F(i-1,j-1)+cost(a_i,b_j)$
- If $a_i$ is aligned with a gap, then the cost is 1 (or whatever the cost of gap is). Hence, the the total cost is $F(i,j) = F(i-1,j)+1$
- If $b_j$ is aligned with a gap, then the cost is 1 (or whatever the cost of gap is). Hence, the the total cost is $F(i,j) = F(i,j-1)+1$
How to know which of the three options to do? We choose the one with minimum cost!
$F(i,j)=min \{F(i-1,j-1)+cost(a_i,b_j), F(i-1,j)+1, F(i,j-1)+1 \}$.
Needleman-Wunsch algorithm
- Compute $F(i,j)$ for every $i,j$ and put in a matrix. First column/row correspond to gap and F(0,0)=0
- As you fill the matrix, keep track of which of the three entries gave you the minimum with an arrow
- Trace back the arrows to construct the alignment (diagonal arrow=nucleotide, vertical/horizontal arrow=gap replacing the nucleotide the arrow is pointing)

Figure 9.3 from Warnow (2018) Computational phylogenetics
In-class example: We want to align $S_1=ATCG$ and $S_2=TCA$ for a cost of substitution of 1 and cost of gap of 2. We will work through the steps of creating the F matrix in class, and then, as homework, you can complete the whole matrix.
How do we get the alignment after building the F matrix?
We trace back the arrows from the bottom right corner:

ATCG
-TCA
Take-home message: The final alignment depends on the costs of gaps and substitutions.
YouTube
You can watch all the steps of the Needleman-Wunsch algoritm in this YouTube video.