Alignment methods (Part 2)
Previous class check-up
- We reviewed the algorithms for pairwise sequence alignments (Needleman-Wunsch algorithm)
Learning objectives
At the end of today’s session, you
- will be able to explain the most widely used algorithm for multiple sequence alignment
Pre-class work
None.
3. Multiple sequence alignment
- The Needleman-Wunsch is the magic algorithm that allows us to align two sequences
- We want to expand the pairwise sequence alignment to multiple sequence alignment
- Progressive alignment: the most widely used algorithm (e.g. ClustalW)
- Iterative refinement algorithm: improvement over progressive alignment by doing sequential alignments until convergence of score (e.g. mafft, muscle)
Progressive alignment
- Compute rooted binary tree (guide tree) from pairwise distances
- Build MSA from the bottom (leaves) up (root)
Stop and check
What is a rooted binary tree?

Figure 9.9 in Warnow (2018) Computational phylogenetics
Progressive alignment algorithm
- Align all pairs of sequences using the Needleman-Wunsch algorithm
- For every pairwise alignment, we calculate its cost based on the cost of gap (e.g. unit cost) and the cost of substitution (e.g. unit cost)
- We estimate the tree from distances: we will learn this in Lecture 8. Let’s pretend we already have the tree
- We build the alignments on the tree from the leaves to the root (bottom-up)
- For the leaves, we build the pairwise alignments for (a,b) and for (d,e) using the Needleman-Wunsch algorithm
- For internal nodes, we need to know how to align alignments
What are the ingredients that we need to know to perform MSA via progressive alignment?
- Perform pairwise sequence alignment via Needleman-Wunsch (check!)
- Calculate the cost of a pairwise sequence alignment (check!)
- Calculate a tree from distances (Lecture 8)
- Perform alignment of alignments (missing)
How to align alignments
We need a new concept called “profile”.
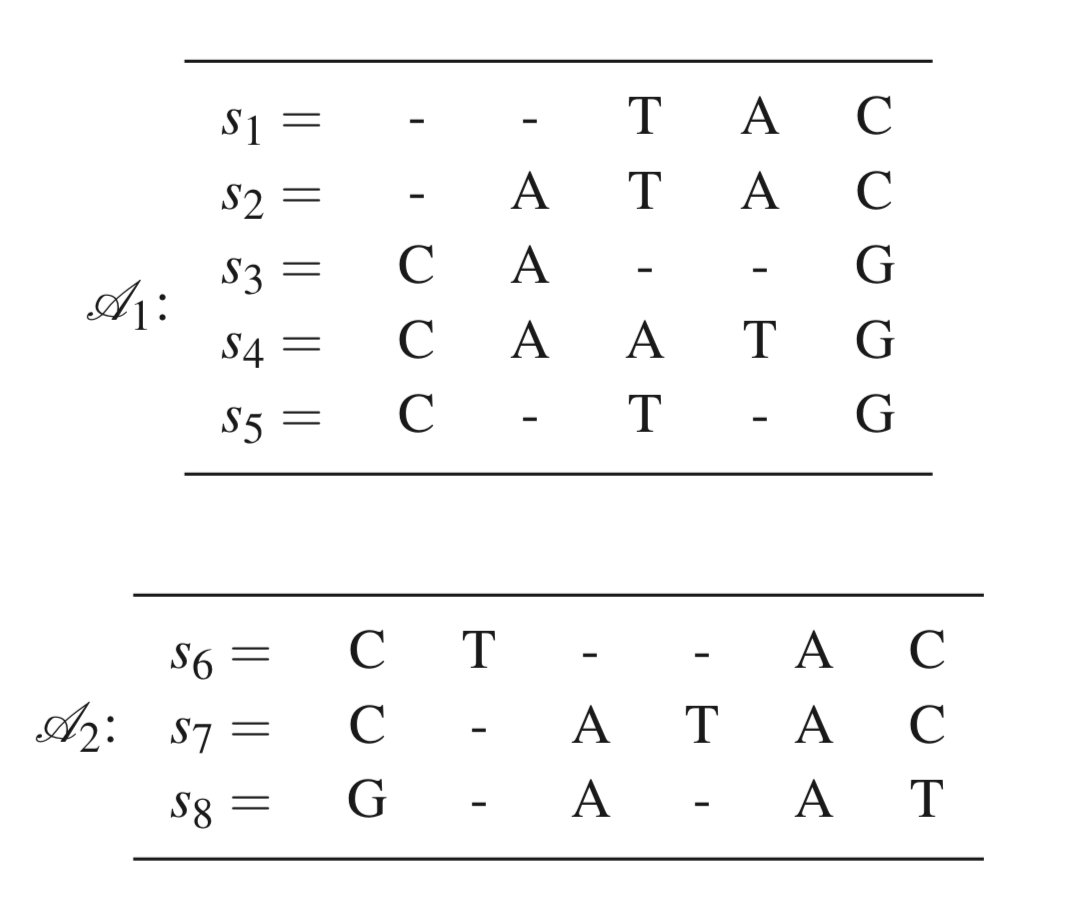

Aligning alignments
- Construct profiles
- Define the cost of putting $a_i, b_j$ together. We want to minimize the expected cost between profiles
- Use Needleman-Wunsch to align $P_1$ and $P_2$ based on the costs
Aligning alignments: defining the costs
Treat $a_i$ in $P_1$ and $b_j$ in $P_2$ as probability models: $P(x \vert a_i)$ is the probability of observing nucleotide $x$ in position $i$ on the profile $P_1$ (Example: What is $P(A \vert a_1)$?)
We define the cost as

In-class exercise
What is the $cost(a_3,b_2)$?
Homework
Instructions: Build the cost matrix for the two following profiles. This means that you want to calculate $cost(a_i,b_j)$ for all $i$ and $j$.
YouTube
You can watch the computation of the cost matrix in this YouTube video. This video has two errors: $cost(a3,b1)=1/4$ instead of 1 and $cost(a4,b6)=7/9$ instead of 8/9.
Aligning the alignments: we have the cost matrix, now what?
Assume we got the following cost matrix
a1 a2 a3 a4 a5
b1 [ 1/3 1 1 1 8/15 ]
b2 [ 1 1 1/4 2/3 1 ]
b3 [ 1 0 3/4 1/3 1 ]
b4 [ 1 1 1/4 2/3 1 ]
b5 [ 1 0 3/4 1/3 1 ]
b6 [ 1/3 1 9/12 8/9 11/15]
and we will use it to align the two profiles $P_1 = a_1 a_2 a_3 a_4 a_5$ and $P_2 = b_1 b_2 b_3 b_4 b_5 b_6$ with Needleman-Wunsch. The cost matrix above provides the costs of substitutions and we assume a cost of gap of 1.
In-class activity
Let’s recall Needleman-Wunsch: we need the $F(i,j)$ matrix and then trace back the alignment. Let’s do here together some of the entries of the $F(i,j)$ matrix.
Homework
Instructions: Finish Needleman-Wunsch on the two profiles.
- Build the F matrix
- Trace back the alignment from the bottom right corner
Solution: You should get the following alignment which we can translate back to the original sequences.


YouTube
You can watch some of the steps in this YouTube video.
MSA key insights Needleman-Wunsch lies at the core of MSA: if we have two sequences, we align them with Needleman-Wunsch; if we have two alignments, we first convert them to profiles, and then align the profiles with Needleman-Wunsch. The final alignment will depend on the assumptions on the cost of substitutions and costs of gaps
Homework recap here.
For next class: Read the paper ClustalW
Pay attention to:
- What does ClustalW assume about sequence relatedness before alignment?
- What is the role of guide trees and why does that matter?
- What do progressive alignment errors imply for downstream analyses?
- What information is encoded in gap penalties and sequence weighting?
- Does ClustalW claim to infer homology, or does it assume it?
- Under what conditions do the authors caution against overinterpretation?